Corresponding lecture
Description
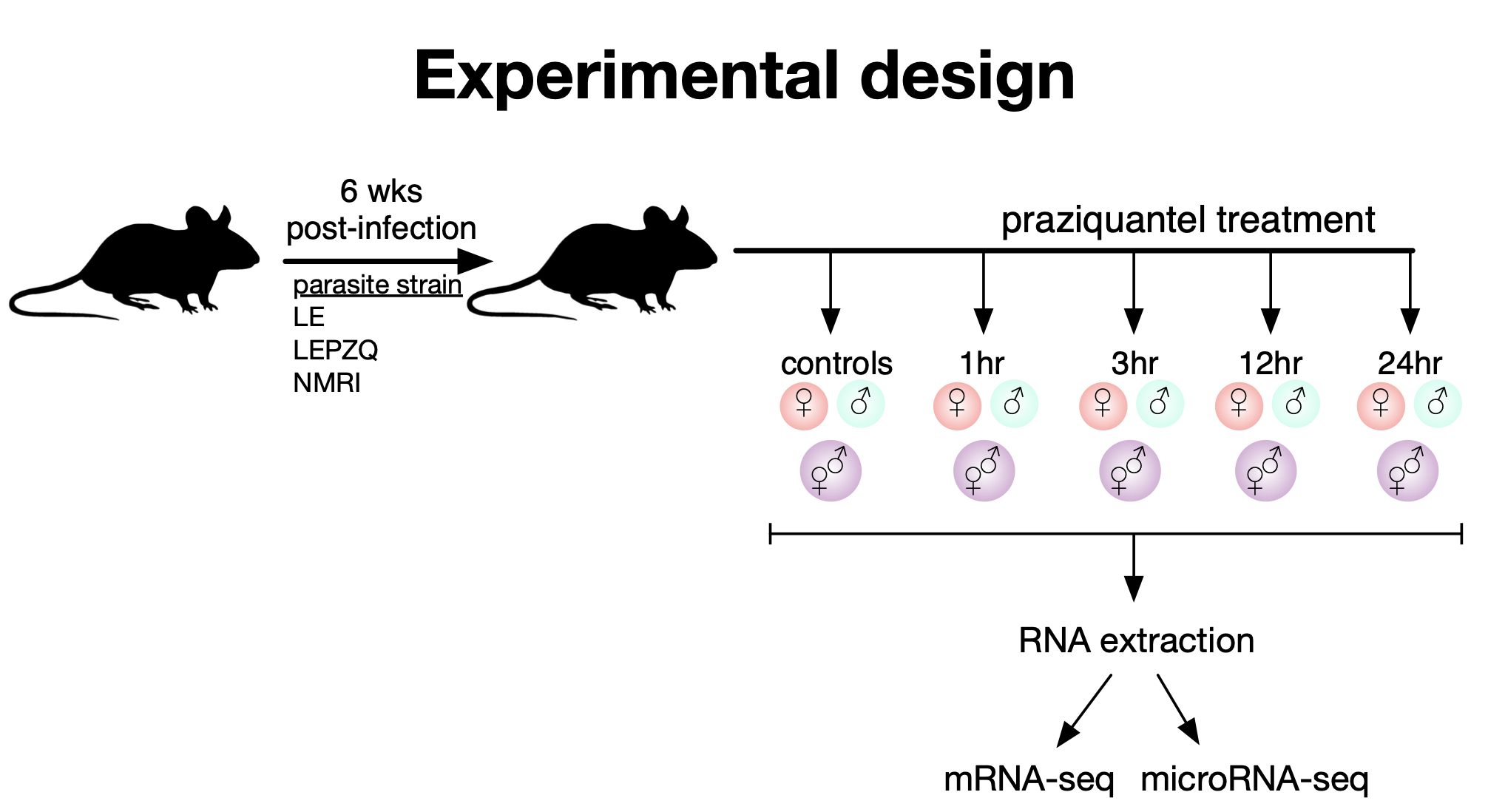
A colleague has asked for your help to mine data produced from a very large RNAseq study of the parasitc worm, Schistosoma mansoni. In this experiment, male (M), female (F), juvenile (J) and mixed sex (X) worms were recovered from infected mice at various timepoints (control, 3hr, 12hr, and 24hr) following in vivo treatment with a low dose of the frontline anti-parasitic drug, praziquantel. Experiments were carried out with three different strains of worms: NMRI, LE, and LEPZQ. In total, 144 samples were sequenced! To begin this challenge, you’ll need to download the Kallisto read mapping results and study design file here and read these data into a new R project.
To succeed in this lab, you will need to use the tools you’ve learned in class for filtering, normalizing, multivariate analysis with PCA, and data wrangling with dplyr.
The first team to submit the most complete answer (via private message on Discord) to the following three questions will win the challenge.
Before you start
- download a fresh copy of the step 1, 2 and 3 scripts from the course website here
- use only the ‘essential’ code chunks from these scripts to complete the challenge.
- don’t try to annotate the data, just import at the transcript level
Part #1
Use plots to show the impact of filtering and normalization on the data.
Tips
- Do not use ‘coord_flip’ in your ggplot code, as the number of samples is so large that horizontal display is preferred
- Don’t use cowplot to patch together your different ggplots into one figure. Again, the number of samples in this particular study makes this too cramped.
Part #2
Which biological variables explain the majority of the variance in this dataset? Please include PCA plot(s) to support your answer?
Tips:
- take advantage of setting the ggplot aesthetics (aes) to map size and shape to different variables from your study design file
Part #3
Using dplyr ‘verbs’, identify the top 10 parasite genes induced by praziquantel treatment in female LE strain worms at the 24hr timepoint compared to control worms. The top 10 genes should be selected and ordered by average Log fold change for the groups of replicates. Please include a table to support your answer.
Bonus points!
Anything interesting stand out to you about the top genes on your list (requires a literature search)?
On your own
If you’re working through this lab on your own, you should be able to complete all three parts above. If you’re an in-person learner and were unable to attend this lab, you should turn in answers to these three parts to the TAs before the start of class next week to get credit.
Solution
script
Download this script to see my answers to the questions above.